Characterization of Gene Expression Differences Resulting from Altered Methyl Donor Abundance
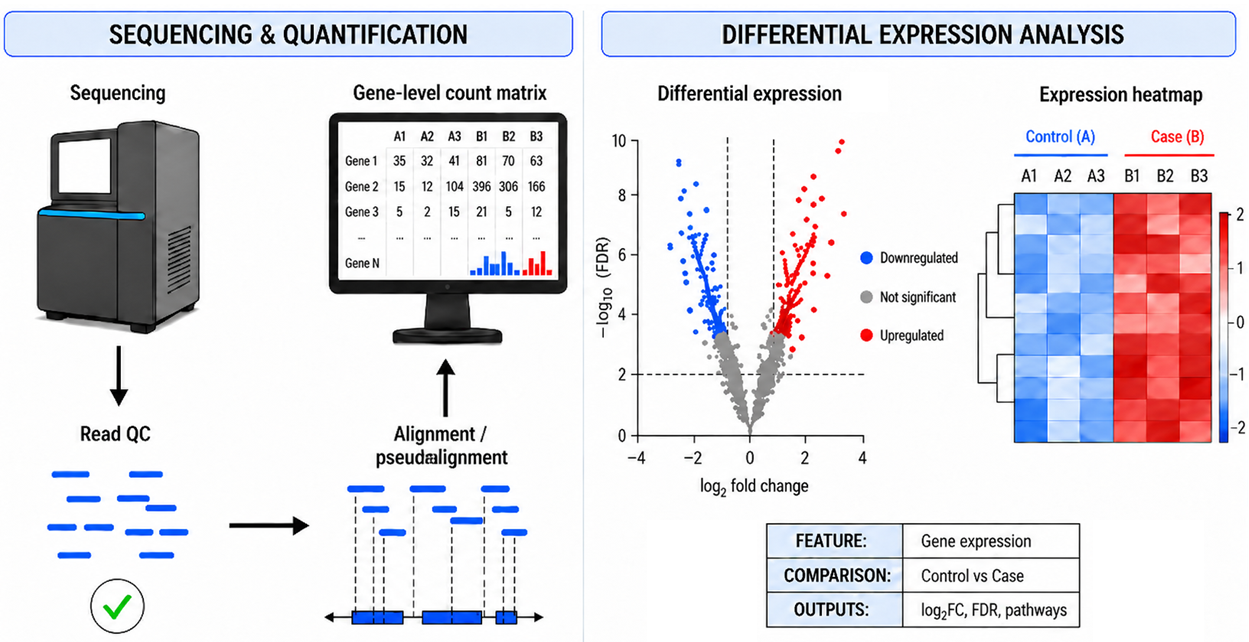
RNA-seq data were analyzed from Saccharomyces cerevisiae strains carrying mutations that alter the availability of S-adenosylmethionine (SAM), the primary cellular methyl donor. The study compared transcriptomic profiles between wild-type yeast and mutant strains to identify gene expression differences and further characterize their biological processes/molecular functions, and pathways affected by altered cellular methylation and methyl cycle capacity.
We developed an RNA-seq analysis workflow utilizing nf-core's RNA-seq pipeline [10.5281/zenodo.1400710] to process raw sequencing reads, perform quality control, and handle alignment and transcript quantification. These outputs were then analyzed with a containerized DESeq2 workflow to identify differentially expressed genes across experimental conditions. Gene Ontology (GO) and pathway enrichment analyses were used to evaluate the broader biological impact of disrupted SAM metabolism.
Deliverables
- Interactive HTML Report: An interactive HTML report summarizing results and including publication-quality graphics, allowing researchers to review, navigate, and contextualize the findings more easily.
- Differential Gene Expression Report: A differential expression report summarizing gene-level expression changes across all experimental comparisons.
- Functional Enrichment Maps: Functional enrichment tables summarizing significantly enriched pathways and Gene Ontology terms for differentially expressed genes.
Publications
- Profiling the compendium of changes in Saccharomyces cerevisiae due to mutations that alter availability of the main methyl donor S-Adenosylmethionine (G3 Bethesda, 2024). DOI: 10.1093/g3journal/jkae002